Download PDF
Inleiding
Jaarlijks wordt in Nederland bij ongeveer 2.000 mensen een kleincellig longcarcinoom (SCLC) gediagnosticeerd; dit betreft ongeveer 15% van alle longkankerdiagnoses. Ten aanzien van behandelingsmogelijkheden wordt er onderscheid gemaakt tussen limited-stage SCLC (SCLC-LS) en extensive-stage SCLC (SCLC-ES). Bij een derde van de patiënten met SCLC is er bij diagnose sprake van LS waarbij de behandeling een curatieve intentie heeft. De standaardbehandeling bestaat uit concurrente chemoradiotherapie waarbij de chemotherapie bestaat uit 4 kuren met een platinumderivaat en etoposide gevolgd door profylactische hersenbestraling. Bij het merendeel van de patiënten is echter binnen 2 jaar sprake van ziekteprogressie en de 5-jaarsoverleving is slechts 29-34%.1 De hier te bespreken ADRIATIC-studie betreft de vergelijking van behandeling met durvalumab, een monoklonaal antilichaam gericht tegen PD-L1, met placebo na standaard chemoradiotherapie bij SCLC-LS.2 EMA heeft behandeling met durvalumab geregistreerd voor patiënten met SCLC-LS die geen progressie hadden na platinumbevattende chemoradiotherapie.
Kankersoort en lijn van behandeling
Adjuvante behandeling met durvalumab na concurrente radiotherapie en chemotherapie met een platinumderivaat en etoposide werd onderzocht bij patiënten met SCLC-LS.
Vergelijking met de referentiebehandeling in Nederland
De in de ADRIATIC-studie gebruikte behandeling bestaande uit chemoradiotherapie met een platinumderivaat en etoposide met een cumulatieve radiotherapiedosis van 60 tot 66 Gy (bij eenmaal daags bestraling) of 45 Gy (bij tweemaal daags bestraling), eventueel gevolgd door profylactische hersenbestraling, komt overeen met de huidige standaardbehandeling van SCLC-LS in Nederland.
Methoden van de studie
De ADRIATIC-studie is een dubbelblind placebogecontroleerde gerandomiseerde fase III-multicenterstudie voor patiënten met een inoperabel stadium I-III histologisch of cytologisch bewezen SCLC-LS die geen progressie hebben na chemoradiotherapie. Om voor inclusie in aanmerking te komen dienden patiënten ten minste 18 jaar te zijn, een WHO-performancestatus van 0 of 1 en een adequate orgaan- en beenmergfunctie te hebben. De belangrijkste exclusiecriteria waren pneumonitis graad 2 of hoger ten gevolge van de chemoradiotherapie, gemengd grootcellig- en kleincelligcarcinoom en een contra-indicatie voor immuuntherapie. Profylactische hersenbestraling was toegestaan.
Randomisatie vond plaats binnen 42 dagen na het voltooien van de chemoradiotherapie, waarbij patiënten 1:1:1 gerandomiseerd werden tussen behandeling met durvalumab en placebo, durvalumab en tremelimumab (een CTLA-4-remmer), of placebo. In november 2020 is na inclusie van de oorspronkelijk geplande 600 patiënten een amendement aangenomen waarbij de inclusie uitgebreid werd met nog 124 patiënten die 1:1 gerandomiseerd werden tussen durvalumab en placebo. Er werden geen patiënten meer geïncludeerd in de durvalumab-tremelimumabgroep. Bij het amendement zijn tevens de eindpunten van de studie aangepast. In dit rapport wordt alleen de vergelijking tussen de durvalumabgroep en de placebogroep besproken. De resultaten van de durvalumab-tremelimumabgroep zijn nog niet gerapporteerd.
Bij de randomisatie werd gestratificeerd naar ziektestadium (I-II versus III) en profylactische hersenbestraling (ja versus nee). De behandeling bestond uit 4-wekelijkse cycli durvalumab 1500 mg of placebo intraveneus toegediend op dag 1 gedurende maximaal 2 jaar. Onderbreking van de therapie was toegestaan. Voor de patiënten in de placebogroep was cross-over naar durvalumab bij progressie niet toegestaan. Data over vervolgbehandelingen werden verzameld.
De uiteindelijke duale primaire eindpunten van de studie waren overleving (OS) en progressievrije overleving (PFS) onafhankelijk en geblindeerd centraal beoordeeld voor de vergelijking tussen de durvalumabgroep en de placebogroep. Secundaire eindpunten waren onder andere PFS na 18 en 24 maanden, OS en PFS in de durvalumab-tremelimumabgroep versus de placebogroep, objectieve responskans, veiligheid en kwaliteit van leven. Responsevaluatie werd gedaan door middel van CT- of MRI-scan bij randomisatie en daarna elke 8 weken gedurende de eerste 72 weken, elke 12 weken tot 96 weken en vervolgens elke 24 weken, totdat er progressie volgens RECIST 1.1 werd vastgesteld. Bijwerkingen werden gegradeerd volgens CTC-AE 4.03.
Kwaliteit van leven werd geëvalueerd middels de vragenlijsten EORTC-QLQ-C30 en QLQ-LC13, die werden afgenomen voor start randomisatie en vervolgens op dag 1 van elke week de eerste 8 weken en daarna elke 4 weken. De vragenlijst EQ-5D-5L werd elke 8 weken afgenomen.
De tweezijdige alfa van 0,05 werd verdeeld tussen de beide primaire eindpunten waarbij een alfa van 0,045 werd toegekend aan OS en 0,005 aan PFS. Wanneer een eindpunt positief was kon de alfa worden hergebruikt voor het testen van het andere primaire eindpunt met een alfa van 0,05. Er waren 348 overlijdens nodig om in een hiërarchische testprocedure met 80% power een verschil in OS vast te kunnen stellen als de hazard ratio (HR) 0,73 is voor de durvalumabgroep ten opzichte van de placebogroep. Met 370 PFS-events heeft de studie een 90% power om een verschil in PFS vast te kunnen stellen met een HR van 0,65 voor de durvalumabgroep ten opzichte van de placebogroep. Er waren ongeveer 724 patiënten nodig, 524 patiënten die werden gerandomiseerd tussen durvalumab monotherapie en placebo (ongeveer 262 patiënten per behandelgroep) en 200 patiënten in de durvalumab-tremelimumabgroep.
Er waren drie interim-analyses gepland, 1 voor PFS en 2 voor OS. De interim-analyse voor PFS was gepland na 308 events. De eerste interim-analyse voor OS was tegelijk gepland met de interim-analyse voor PFS bij een geschat aantal van ongeveer 242 overlijdens en de tweede interim-analyse voor OS zou plaatsvinden na ongeveer 299 overlijdens. Het tweezijdige significantieniveau voor de toetsing van OS bij de hier gerapporteerde eerste interim-analyse was 0,01679 en voor PFS was de grenswaarde voor significantie 0,02805 omdat de analyse voor OS positief was.
Effectiviteit van de behandeling afgezet tegen de bijwerkingen en impact van de behandeling
Tussen september 2018 en augustus 2021 werden 939 patiënten gescreend en 730 patiënten geïncludeerd door 179 centra in Azië, Europa, Noord- en Zuid-Amerika; 264 patiënten in de durvalumabgroep, 266 in de placebogroep en 200 in de durvalumab-tremelimumabgroep. De resultaten van de 530 patiënten uit durvalumabgroep en de placebogroep worden hier besproken. De mediane follow-upduur ten tijde van deze eerste interim-analyse bedroeg 37,2 maanden. De uitgangskarakteristieken in de beide behandelgroepen waren vergelijkbaar: de mediane leeftijd was 62 jaar, de meeste patiënten waren man (69%), voormalig roker (69%) of actuele roker (22%), met stadium III ziekte (87%) bij diagnose. Het platinumderivaat bij de chemoradiotherapie bestond voor 66% uit cisplatine en 88% voltooide 4 kuren chemotherapie. De radiotherapieschema’s die werden gebruikt, waren eenmaaldaagse radiotherapie bij 72% van de patiënten en gehyperfractioneerde tweemaaldaagse radiotherapie bij 28%, waarbij 94% van de patiënten de aanbevolen radiotherapiedosis ontving van 60 tot 66 Gy eenmaal daags of 45 Gy tweemaal daags en 54% van de patiënten kreeg profylactische hersenbestraling.
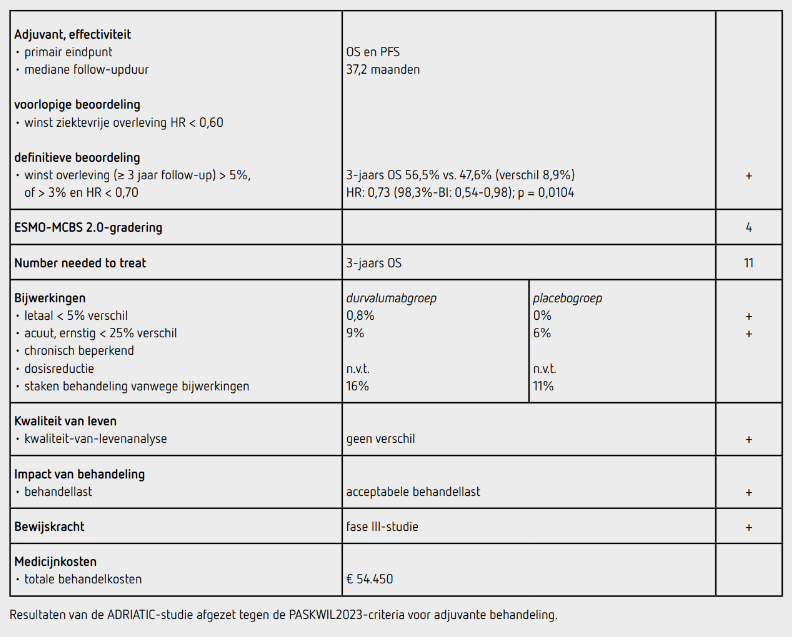
Ten tijde van de data-cutoff waren 261 patiënten overleden. Het primaire eindpunt OS was significant langer in de durvalumabgroep dan in de placebogroep (HR: 0,73 [98,3%-BI: 0,54-0,98]; p = 0,0104). De mediane OS bedroeg 55,9 maanden (95%-BI: 37,3-niet bereikt) in de durvalumabgroep en 33,4 maanden (95%-BI: 25,5-39,9) in de placebogroep. De 2-jaars OS was 68,0% in de durvalumabgroep en 58,5% in de placebogroep en de 3-jaars OS was respectievelijk 56,5% en 47,6%. Bij de analyse van PFS hadden 308 patiënten ziekteprogressie of waren overleden. Het primaire eindpunt PFS was significant langer in de durvalumabgroep dan in de placebogroep (HR: 0,76 [97,2%-BI: 0,59-0,98]; p = 0,0161). De mediane PFS bedroeg 16,6 maanden (95%-BI: 10,2-28,2) in de durvalumabgroep en 9,2 maanden (95%-BI: 7,4-12,9) in de placebogroep. In de vooraf gespecificeerde subgroepen was de HR voor OS en PFS consistent in het voordeel van de durvalumabgroep.
Van de 262 patiënten (99,2%) in de durvalumabgroep en de 265 patiënten (99,6%) in de placebogroep die ten minste 1 dosis durvalumab of placebo ontvingen, voltooiden respectievelijk 88 patiënten (34%) en 70 patiënten (26%) de maximaal toegestane 24 maanden behandeling. Aan beide groepen werd mediaan 9 kuren gegeven. In de durvalumabgroep werd de behandeling onderbroken vanwege bijwerkingen bij 91 patiënten (35%) en 43 patiënten (16%) stopten de behandeling vanwege bijwerkingen. In de placebogroep werd de behandeling onderbroken bij 76 patiënten (29%) en stopten 28 patiënten (11%) vanwege bijwerkingen met de behandeling.
Behandelingsgerelateerde bijwerkingen van graad 3 of hoger traden op bij 23 patiënten (9%) in de durvalumabgroep en bij 16 patiënten (6%) in de placebogroep. Immuungemedieerde bijwerkingen kwamen voor bij 84 patiënten (32%) in de durvalumabgroep en bij 27 patiënten (10%) in de placebogroep, maar waren meestal van graad 1 of 2. Er zijn 2 patiënten (1%) in de durvalumabgroep overleden als gevolg van een bijwerking (encephalopathie en pneumonitis) en er zijn geen patiënten in de placebogroep overleden door bijwerkingen van de behandeling. Er was geen verschil in kwaliteit van leven tussen de durvalumabgroep en de placebogroep gedurende de eerste 2 jaar.3 Na progressie hebben 95 patiënten (36%) in de durvalumabgroep en 127 patiënten (48%) in de placebogroep een vervolgbehandeling gekregen, waarbij respectievelijk 82 (31%) en 114 patiënten (43%) chemotherapie, en 17 (6%) en 31 patiënten (12%) immuuntherapie als onderdeel van hun volgende lijn therapie ontvingen.
Kwaliteit van de studie en interpretatie van de uitkomsten
De gebruikte behandeling bestaande uit concurrente chemoradiotherapie met een platinumderivaat en etoposide gevolgd door optioneel profylactische hersenbestraling is conform het Nederlandse behandellandschap voor SCLC-LS waarbij geen adjuvante behandeling wordt gegeven. In de ADRIATIC-studie kwam 39% van de patiënten uit Europa. SCLC wordt vaker gezien bij mannen dan vrouwen, al is 69% man wel aan de hoge kant in deze studie. Verreweg het grootste deel was roker of ex-roker, wat te verwachten is bij dit type kanker. Mogelijk omdat OS het (duale) primaire eindpunt was, is ervoor gekozen cross-over in de studie niet toe te staan, hetgeen wel invloed kan hebben op dit eindpunt. Een vervolgbehandeling werd gegeven aan circa 80% van de patiënten met progressie in beide groepen wat bij de meesten bestond uit chemotherapie. Slechts een klein percentage van de patiënten in de placebogroep heeft na progressie alsnog immuuntherapie gekregen. Bij een amendement in november 2020 is de opzet van de studie aanzienlijk gewijzigd.
Aanvankelijk waren de primaire eindpunten PFS voor de vergelijking tussen de durvalumab-tremelimumabgroep en de placebogroep en PFS voor de vergelijking tussen de durvalumabgroep en de placebogroep. Bij het amendement is PFS voor de vergelijking durvalumab-tremelimumab versus placebo een secundair eindpunt geworden en is OS voor de vergelijking durvalumab versus placebo als (duaal) primair eindpunt toegevoegd. Het aantal te includeren patiënten in de durvalumabgroep en placebogroep is verhoogd. De reden die de auteurs geven voor deze wijzigingen zijn de resultaten van de CASPIAN-studie waarbij er geen meerwaarde bleek te zijn van toevoegen van tremelimumab aan durvalumab bij SCLC-ES. In deze studie werd wel OS-winst aangetoond van durvalumab ten opzichte van placebo.4 Resultaten van de durvalumab-tremelimumabgroep van de ADRIATIC-studie zijn tot op heden niet bekend.
Discussie
In de ADRIATIC-studie wordt een statistisch significant voordeel aangetoond voor het primaire eindpunt OS van adjuvante behandeling met durvalumab ten opzichte van placebo na chemoradiotherapie bij SCLC-LS (HR: 0,73 [98,3%-BI: 0,54- 0,98]; p = 0,0104). De mediane follow-upduur bij deze interim-analyse is 37 maanden waarbij een verschil in 3-jaars OS van 8,9% wordt gezien. Deze uitkomsten voldoen aan de criteria voor een positief advies volgens de PASKWIL2023-criteria voor adjuvante behandeling. Ondanks de ongunstige prognose van SCLC, ook bij LS, is lokale behandeling met chemoradiatie de hoeksteen van de therapie en heeft deze behandeling een in opzet curatieve intentie. Om deze reden zijn de adjuvante PASKWIL-criteria van toepassing.
Ook de PFS laat een significant voordeel voor durvalumab zien (HR: 0,76 [97,2%-BI: 0,59-0,98]; p = 0,0161). Zoals hierboven omschreven is er een belangrijk amendement geweest in november 2020. Het is echter niet onze inschatting dat dit de betrouwbaarheid van de resultaten voor het effect van durvalumab versus placebo in belangrijke mate beïnvloed heeft. Slechts een derde van de patiënten heeft de maximale behandelduur van 2 jaar voltooid. De duur van deze behandeling is lang en een goede biologische onderbouwing ontbreekt. Het brengt ook aanzienlijke kosten met zich mee. In het algemeen werd de behandeling goed verdragen. Slechts een klein deel van de patiënten die bij progressie in de placebogroep een vervolgbehandeling kreeg, ontving immuuntherapie. Hierdoor blijft onduidelijk wat de winst is van immuuntherapie in de adjuvante setting na chemoradiotherapie vergeleken met toepassing bij optreden van recidief.
Kosten
De kosten van behandeling met durvalumab voor een cyclus van 28 dagen zijn 6.050 euro. Bij een behandeling met mediaan 9 cycli zijn de totale kosten 54.450 euro (bron: medicijnkosten.nl d.d. oktober 2025).
Conclusie
In de hier besproken ADRIATIC-studie wordt bij patiënten met SCLC-LS na een mediane follow-upduur van 37,2 maanden een statistisch significant langere OS gezien na adjuvante behandeling met durvalumab in vergelijking met placebo na chemoradiotherapie (HR: 0,73 [98,3%-BI: 0,54-0,98]; p = 0,0104). Het verschil in 3-jaars OS bedraagt 8,9%. Deze resultaten voldoen aan de criteria voor een positief advies volgens de PASKWIL2023-criteria voor adjuvante behandeling.
Referenties
- Faivre-Finn C, Snee M, Ashcroft L, et al. Concurrent once-daily versus twice-daily chemoradiotherapy in patients with limited-stage small-cell lung cancer (CONVERT): an open-label, phase 3, randomised, superiority trial. Lancet Oncol 2017;18(8):1116-25.
- Cheng Y, Spigel DR, Cho BCet al. Durvalumab after chemoradiotherapy in limited-stage small-cell lung cancer. N Engl J Med 2024;391(14):1313-27.
- Novello S, Cheng Y, Spigel D et al. Patient-reported outcomes (PROs) with consolidation durvalumab versus placebo following cCRT in limited-stage SCLC: ADRIATIC. Journal of Thoracic Oncology 2024;19(10, Supplement):S124-5.
- Goldman JW, Dvorkin M, Chen Y et al. Durvalumab, with or without tremelimumab, plus platinum-etoposide versus platinum-etoposide alone in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): updated results from a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2021;22(1):51-65.